May 31, 2016 - by Simone Ulmer
Extreme temperature and pressure conditions prevail at hydrothermal vents in the deep ocean. Adding to that the distinct structuring of the vents resulting from sulphide-rich minerals and the lack of light, scientists see parallels with the early Earth: an ideal breeding ground for the chemical evolution of organic molecules, the building blocks of life. Now researchers from Hungary, Switzerland and Italy using ab initio computational methods on "Piz Daint" supercomputer have for the first time specifically simulated the reaction chain that enables the formation of ammonia (NH3), one of the prebiotic molecules that are key to the origin of life. The results support the theory of chemoautotrophic development of life, according to the researchers. Detailed knowledge of the reaction process can also provide information about the reaction behaviour and oxidation of minerals, and the catalytic properties of their surfaces. This is of great interest for materials research.
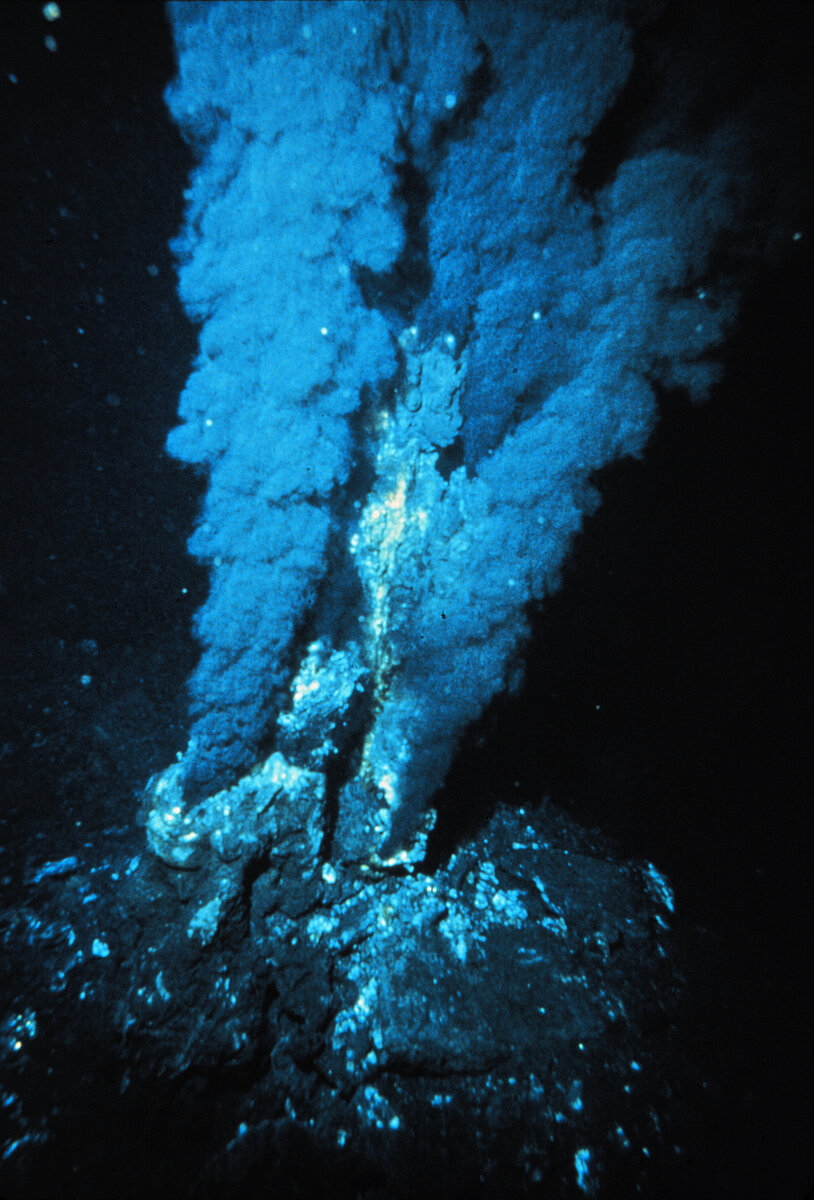
Black smoker as the prebiotic soup
‘Black smokers’ form in volcanically and tectonically active areas of the deep ocean floor. Seawater penetrating through cracks and fissures hundreds of meters deep into the oceanic crust is heated to several hundred degrees Celsius. On its way through the crust the water dissolves various iron and manganese-containing minerals, thus becoming enriched with their sulphides and salts. Hot seawater re-emerging from the crust reacts with the ice-cold water of the deep ocean: black smoke may form through precipitation of iron sulphide in the form of the mineral pyrite; in this way the vent can grow several metres tall.
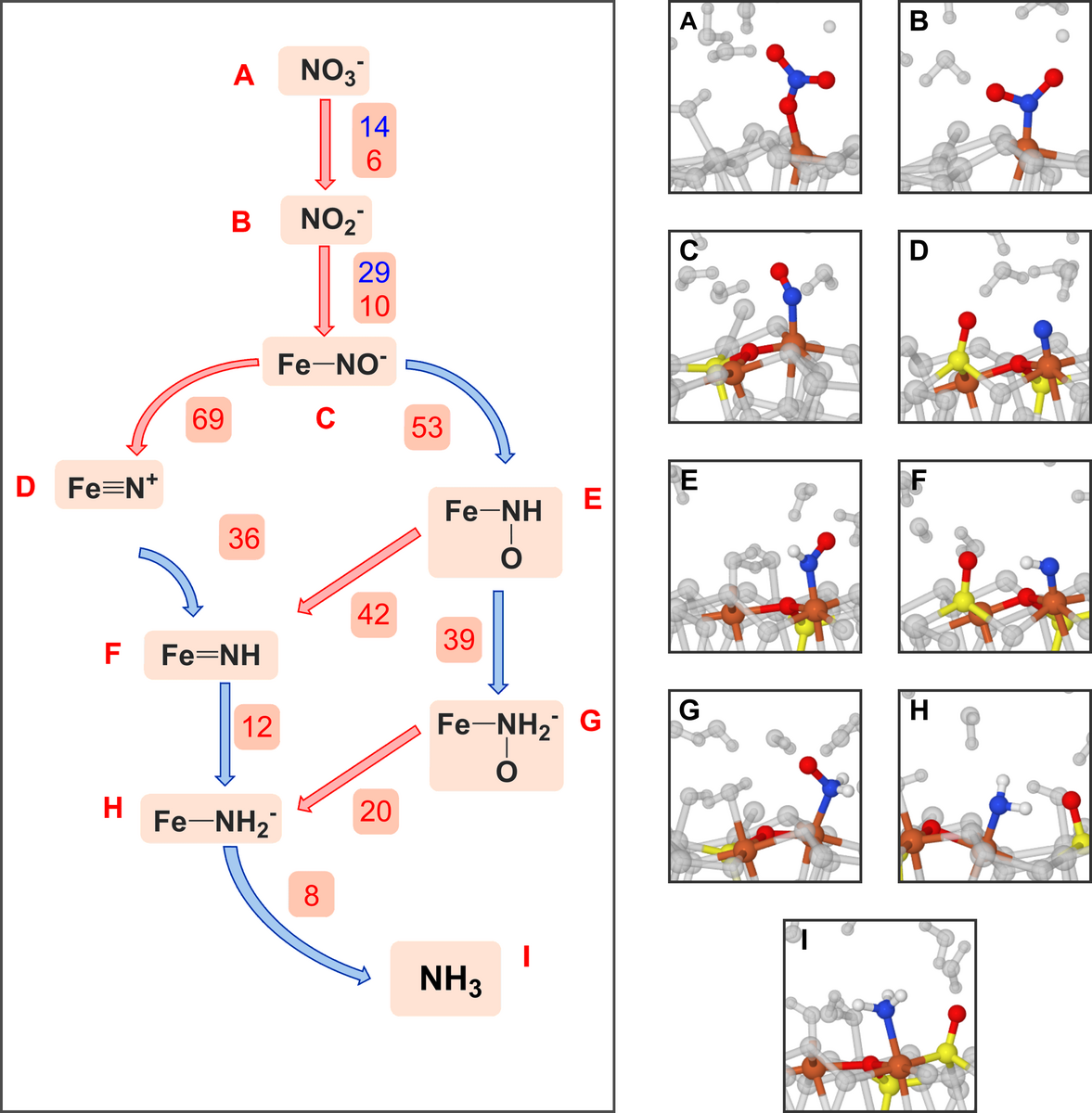
In the late nineties scientists experimentally simulated these extreme conditions in order to study the processes as they unfold. They found that the corresponding iron sulphides can catalyse the reduction of nitrate anions (NO3-) to ammonia (NH3). However, the reaction mechanism – the details of what happens along the reaction path from the starting material to the final product – has so far remained unclear. But now, researchers led by Andras Stirling at the Research Centre for Natural Sciences of the Hungary Academy of Sciences and Matthias Krack at the Paul Scherrer Institute in Villigen have simulated this reaction path on ‘Piz Daint’ using ab initio computational methods. The researchers involved in the study were all formerly in the research group under ETH Professor Michele Parrinello. Parinello’s group conceptualised the simulation of prebiotic processes on iron sulphide surfaces and developed the metadynamics simulation technique in use here.
“Metadynamics simulations allow us to explore complex chemical or physical processes such as protein folding, phase transitions or changes in crystalline structure, which usually cannot be observed on the time scale of normal molecular dynamics simulations”, says Krack. These processes must overcome an energy barrier (activation energy) that does not reveal itself in experimental observations. Often therefore, assistance from supercomputers is the only way to gain detailed knowledge of highly complex chemical reactions, as these take place very rapidly and not all of the intermediate steps are observable by experiment.
Tracing the reaction steps
This is why the researchers used metadynamics computations to investigate the individual reaction steps that lead to ammonia forming on the pyrite mineral surface. Calculation results are rendered as images; from these ‘flip books’ it is possible to reproduce the movements of individual atoms and the way they react together chemically. It is these chemical reactions which most interest the researchers, because from them potential pathways can be deduced. Proceeding from the calculated activation energies of the reactions the researchers can ultimately determine the most likely course, because chemical reactions prefer the lowest-energy pathways.
According to the researchers, the simulations show that nitrate anions on pyrite can be converted to ammonia step by step through the transfer of individual atoms, whereby the pyrite surface is a major participant in the reaction. The mineral assists the process by binding the nitrogen-containing reagents to itself while the chemical reaction is taking place. It is thus responsible both for encouraging the reaction to complete and for facilitating it in the first place.
However clear the picture appears now, it was quite challenging to choose the most important reaction paths and design the simulations so that nothing significant was missing, explains Stirling. Without a doubt, the metadynamics simulations were only possible through efficient implementation of the ab initio computational methods within the framework of the CP2K program package and the availability of the ‘Piz Daint’ high performance computer.
Reference
Stirling A, Rozgonyi T, Krack M & Bernasconi M: Prebiotic NH3 Formation: Insights from Simulations, Inorganic Chemistry (2016), 55, 1934-1939, DOI: 10.1021/acs.inorgchem.5b02911.