
May 08, 2023 – by Simone Ulmer
Theoretical chemist Sandra Luber wants to know exactly what is going on: How do electrons behave in a molecule when it is excited with a laser pulse and made to oscillate? In experimental setups, researchers measure the energy spectra of the excited electrons with a detector and thereby obtain an electronic adsorption spectrum of the molecule, for example. But what happens to the electrons in the time between the laser pulse and the resulting spectrogram remains hidden — only supercomputers like "Piz Daint" can make that visible.
Calculating such dynamic processes in spectroscopy is time-consuming and cost-intensive, which means that even world-class supercomputers can only simulate small systems. However, Luber, a professor of theoretical chemistry at the University of Zurich, together with her PhD student Ruocheng Han and postdoc Johann Mattiat, recently presented an algorithm in Nature Communications that works ten times faster, and without sacrificing accuracy.
Observing electron dynamics in real time
Luber and her team employed “Piz Daint” to model the excited molecules’ dynamics, including the quantum mechanical states. To do this, they used software packages such as CP2K that contain methods for calculating the quantum mechanical states in the atom or molecule in real time. This enabled them to follow how the wave function of the electrons changed over time after the laser pulse. Most importantly, they could see how the higher energy levels induced by the laser are occupied by the electrons and could take "detailed pictures" of the molecule at any point in the experiment. "This helps us analyse the structure and dynamics of a molecule," said Luber.
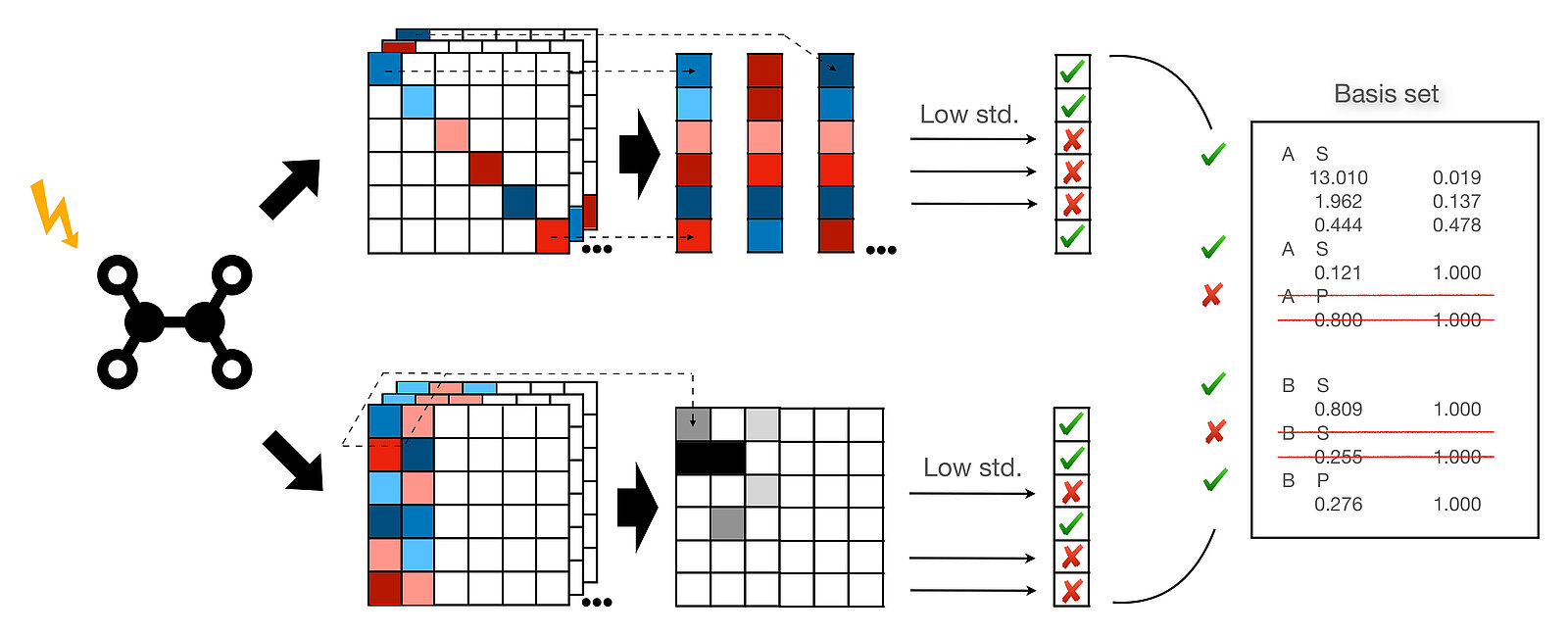
In order to avoid trial and error, the researchers ideally wanted to develop an automated method for speeding up these calculations. Specifically, the algorithm that the team created now optimises the so-called basis sets of functions that then CP2K, for example, uses for the calculations. The team achieved this by identifying two indicators: one indicator that can be used to capture the importance of each basis function for calculating the spectrum; and another indicator that provides information about how important they are for correctly tracking the quantum mechanical states over time.
Using "Piz Daint", the researchers tested their new algorithm on various molecules, ranging from molecular hydrogen and water to a silver cluster and zinc phthalocyanine among other important molecules for industry. With the new algorithm, the researchers reached their goal faster and with the same precision, as comparisons of the simulated absorption spectra with conventionally modelled spectra showed. All other quantum mechanical programmes besides CP2K that also use atom-centered basis sets could use the new procedure, Luber said.
Different functions for different areas in the atom
Optimised basis sets already exist for calculations of molecules mainly in the ground state. "However, such special basis sets for the simulation of excited molecular states did not exist until now," Luber emphasised. "What's more, our newly generated basis sets are even system and environment specific." The researchers made this surprising discovery during test simulations of silver atoms within silver clusters, which have different chemical properties depending on the symmetry and environment in the cluster. "We could observe that our algorithm even finds different basis sets for these different silver atoms," said Luber.
This means that the algorithm distinguishes the environments in the molecule: If, for instance, the polarization of the electron density is important, the algorithm adds polarization functions; for larger distances from the atom it adds diffuse functions. "We can see which type of function is important in which area of the atom or molecule. This gives us a lot of additional information about the molecule’s chemistry." Luber and her team have thus come a lot closer to their goal of knowing exactly what is going on in the excited molecules.
Image above: Adobe stock/arthead
Reference:
Han R, Mattiat J & Luber S: Automatic purpose-driven basis set truncation for time-dependent Hartree–Fock and density-functional theory. Nature Communications (2023). https://doi.org/10.1038/s41467-022-35694-4
The image of the schematic diagram of the basis set truncation is under Open-Access Licence: https://creativecommons.org/licenses/by/4.0/. Find more information about the figure here: https://www.nature.com/articles/s41467-022-35694-4/figures/1
This article may be used on other media and online portals provided the copyright conditions are observed.