September 09, 2021 - by Santina Russo
When scientists were able to start sequencing DNA, it marked the beginning of a revolution for biological research — a revolution fueled by faster next-generation methods, which soon led to the mapping of whole genomes. These DNA sequencing techniques worked at the level of one specific cell and one specific DNA molecule, profoundly advancing the understanding of biological processes and boosting medical precision diagnostics. For proteins, however, such a breadth of view is still out of reach.
Today’s methods for identifying a protein’s sequence are based on mass spectrometry. They do work well for purified protein samples and simple mixtures, but they fall short when it comes to the complex mixture of an entire cell comprising a variety of protein forms of diverse segment composition, hereditary variants and post-translational modifications. Also, since individual protein molecules can not be replicated in the lab like DNA strands, they can’t be sequenced individually — yet. Scientists worldwide are working on approaches to tackle this gap and enable the sequencing of the proteins at single-cell and ultimately at single-molecule level. Among them is Mauro Chinappi, an associate professor in Fluid Dynamics at the University of Rome Tor Vergata.
Complex proteins
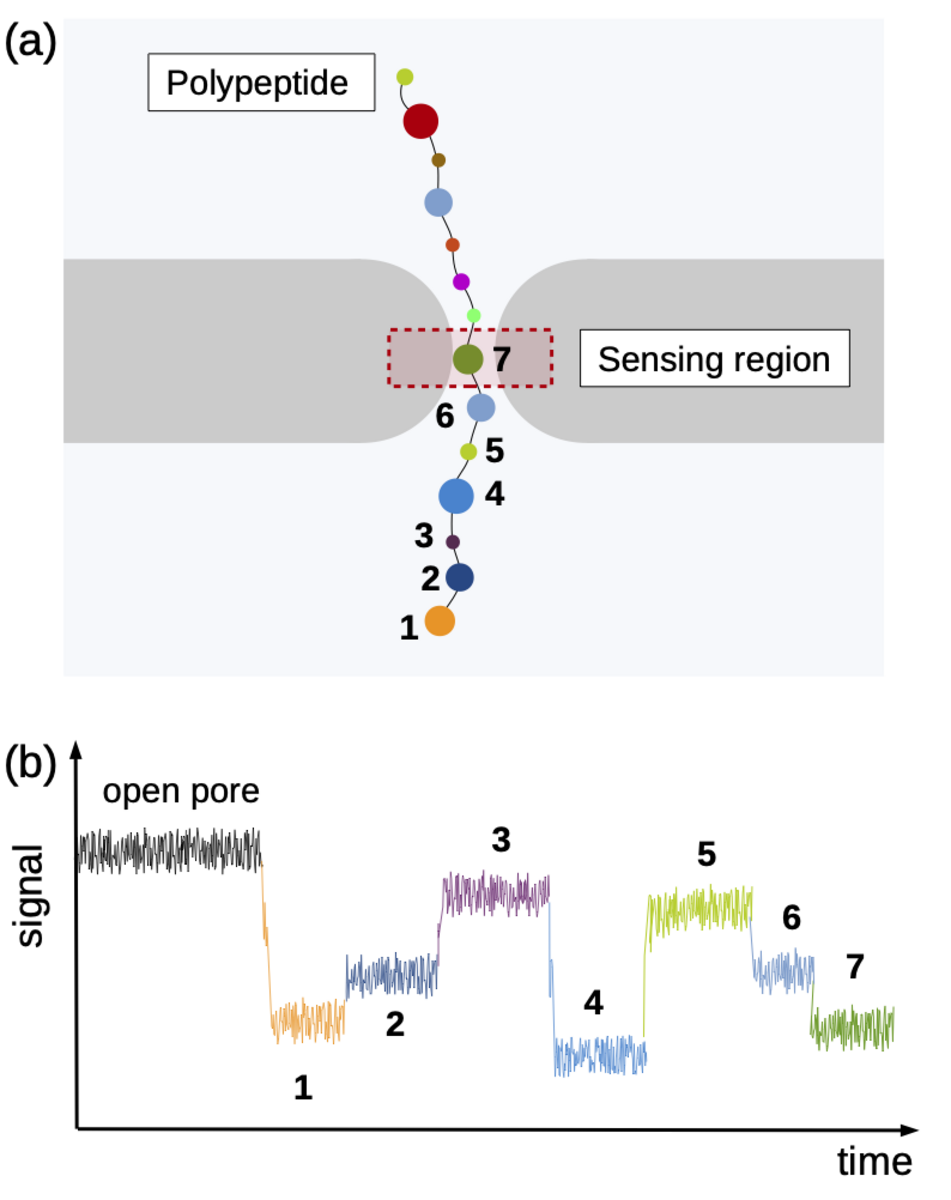
Using the “Piz Daint” supercomputer at CSCS, Chinappi and his co-workers explored a promising approach that employs biological nanopores. These channel proteins are embedded in cell membranes and provide a passageway in or out of the cell. If an electric voltage is applied, such pores provide passage for a steady ion flow. They can be used as molecular sensors based on the measureable drop in ion current that occurs when the pore’s most narrow section is blocked by a molecule. This handy property can be exploited when sequencing by channelling a polymer chain through the nanopore bit by bit. Along the way, ideally, each polymer building block creates a slightly different ion current signal by which it can be identified — a nucleotide in a DNA strand, for instance, or, in the case of proteins and peptides, an amino acid.
“This method already works well for DNA sequencing,” says Chinappi. “However, proteins are more complicated than DNA, because they consist of a higher number of individual building blocks. Instead of four different nucleotides, the method must distinguish 20 different amino acids. And, while all nucleotides are negatively charged, amino acids are more varied: Some are negatively charged, some positively charged, and some neutral.”
Identifying the sequence To explore how well different amino acids can be distinguished by nanopore sensing, Chinappi and his team performed an extensive set of molecular dynamics simulations. They used α-hemolysin, one of the most widely studied pores in nanopore sensing, and calculated the ion current blockage generated by four different polypeptide chains, each composed of one amino acid: alanine (Ala), phenylalanine (Phe), tryptophan (Trp) and glutamine (Gln). Based on these results, the team went on to develop a method to estimate the ion current for all 20 amino acids found in proteins.
The results are promising: The nanopore produced different signals for the four investigated amino acids. As expected, larger amino acid residues resulted in a lower ion current. “Also, interestingly, the measured ion current signal reflected differences in hydrophobicity and net charge,” recounts Chinappi. Charged residues left more room for ion movement and resulted in a larger ion signal than uncharged ones of similar volume.
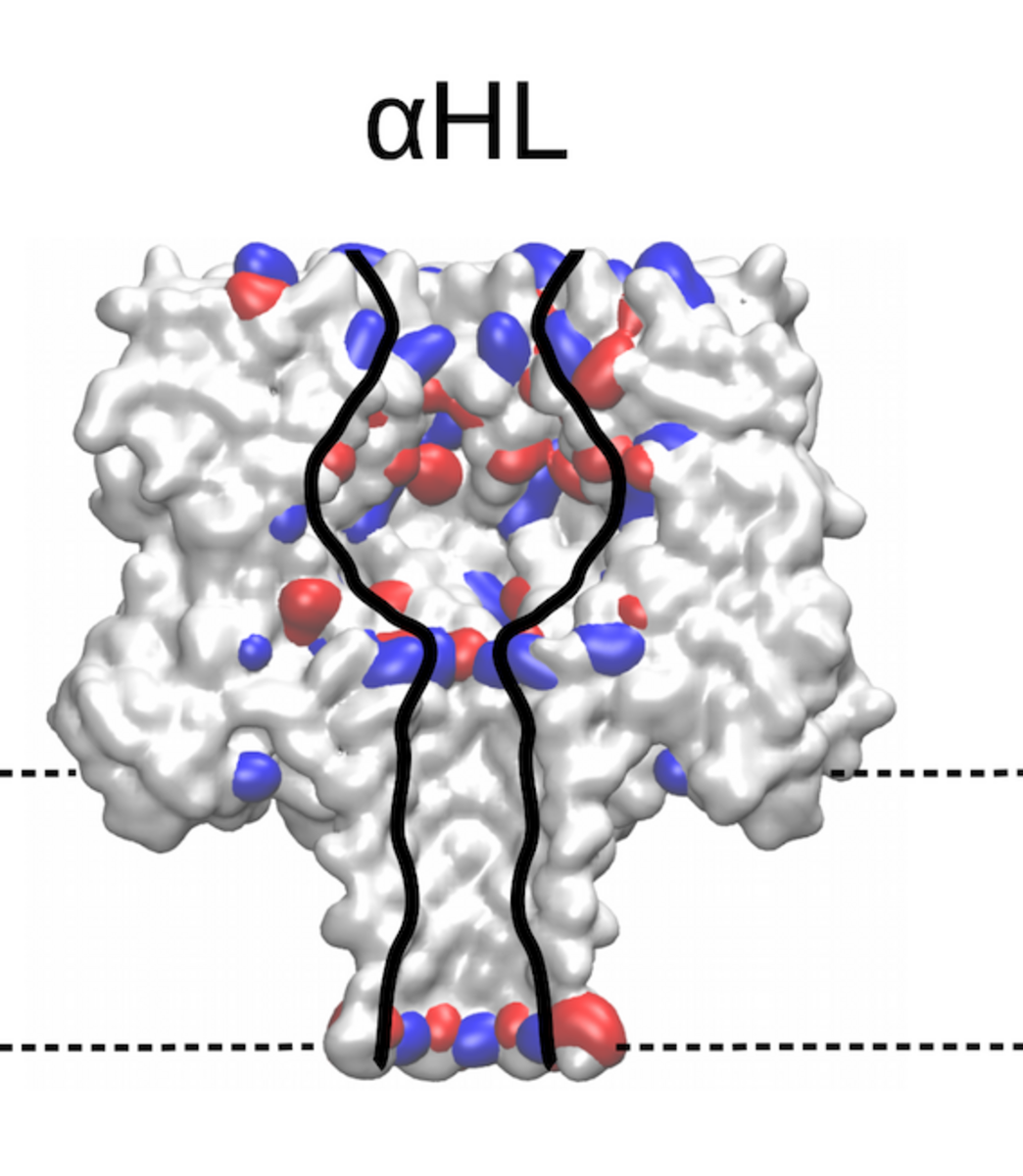
“From the methodological point of view, this means that simple models that assume that nanopore sensing is purely dependent on the blocking residue’s volume will not produce accurate results,” explains Chinappi. The results also indicate that α-hemolysin is indeed potentially able to distinguish among the different amino acid residues — certainly between some amino acids and some classes of amino acids.
Still, for some residues with similar volume, the pore blocking is rather close and therefore also the ion current signal. “For now, we are not quite able to distinguish all 20 amino acids yet,” says Chinappi. In their simulations, however, he and his co-workers identified locations in α-hemolysin that could be engineered towards better distinguishability, particularly a second narrowing in the pore that masks an accurate amino acid identification.
Bringing in the protein
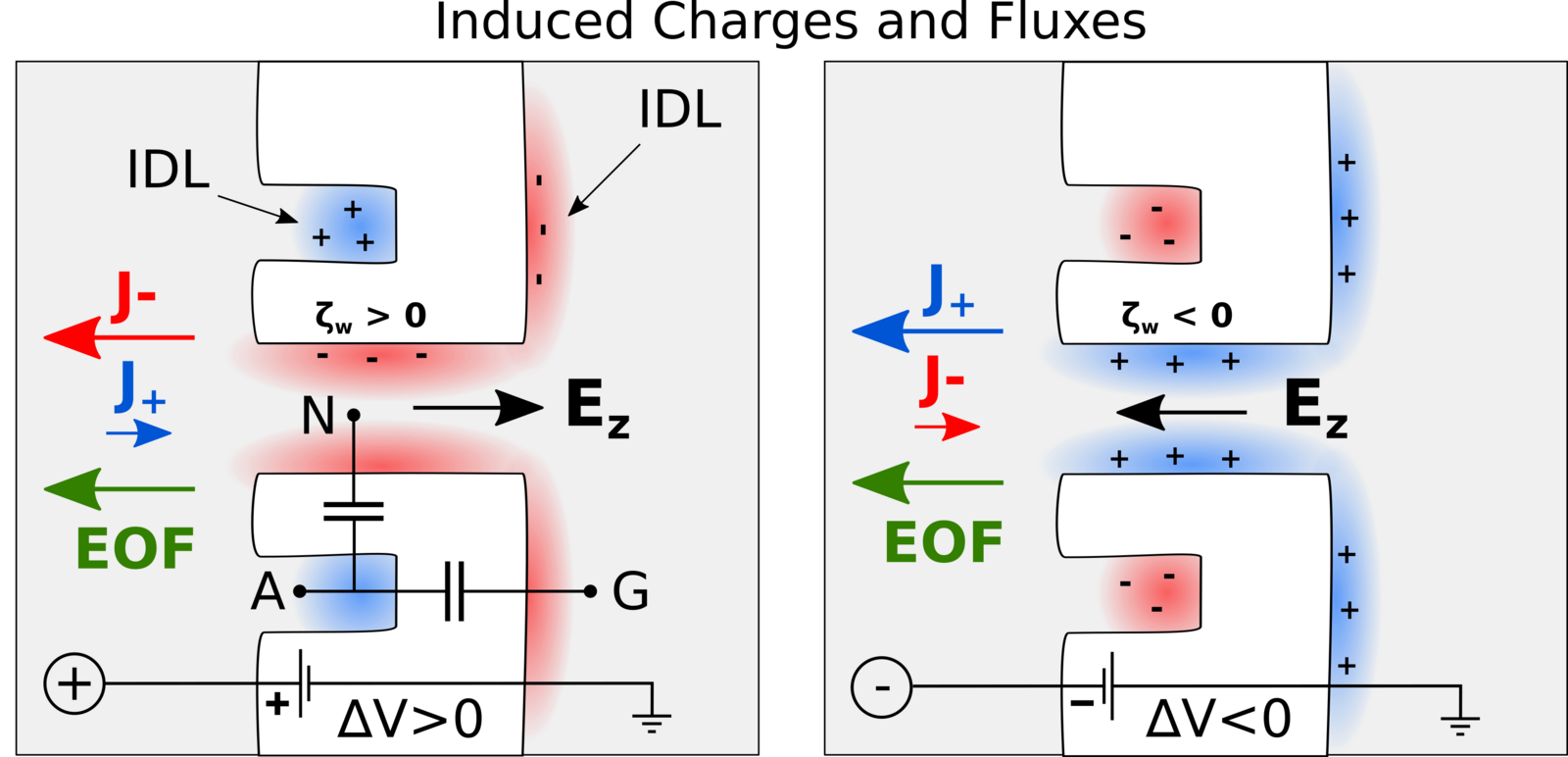
Besides the actual identification of amino acids, another challenge is how to bring the protein molecules to the nanopore in the first place. Chinappi’s team recently explored if this transport can be achived by so-called electroosmosis, in which electric forces induce a water flow that ideally brings the protein to the pore. As was known before, such a directed electroosmotic flow can be achieved through a net positive or negative charge on the central internal surface of the pore. This induces a selective motion of oppositively charged ions, and these, in turn, bring along a number of water molecules. The result is an electroosmotic water flow that can direct peptides and proteins to where they should be — in this case into the nanopore.
However, this way of inducing a electoosmosis has a disadvantage: It is dependent on the internal surface in the pore and thus on the same spot that is responsible for its sensing capabilities. “This means that if you aim to engineer the pore’s capabilities to capture protein molecules, you will also affect its capability to identify amino acids, and vice versa,” explains Chinappi.
Undeterred, he and his co-workers have already begun looking for another way to induce an electroosmotic flow. In their molecular dynamics simulations, they probed a nanopore with an additionally introduced cavity on its exterior that attracts ions. Placed properly, this ion trap induces a complementary current of oppositively charged ions through the pore, as the results showed — and therefore, again, a directed water flow that can bring proteins to the pore.
Towards engineering the perfect nanopore
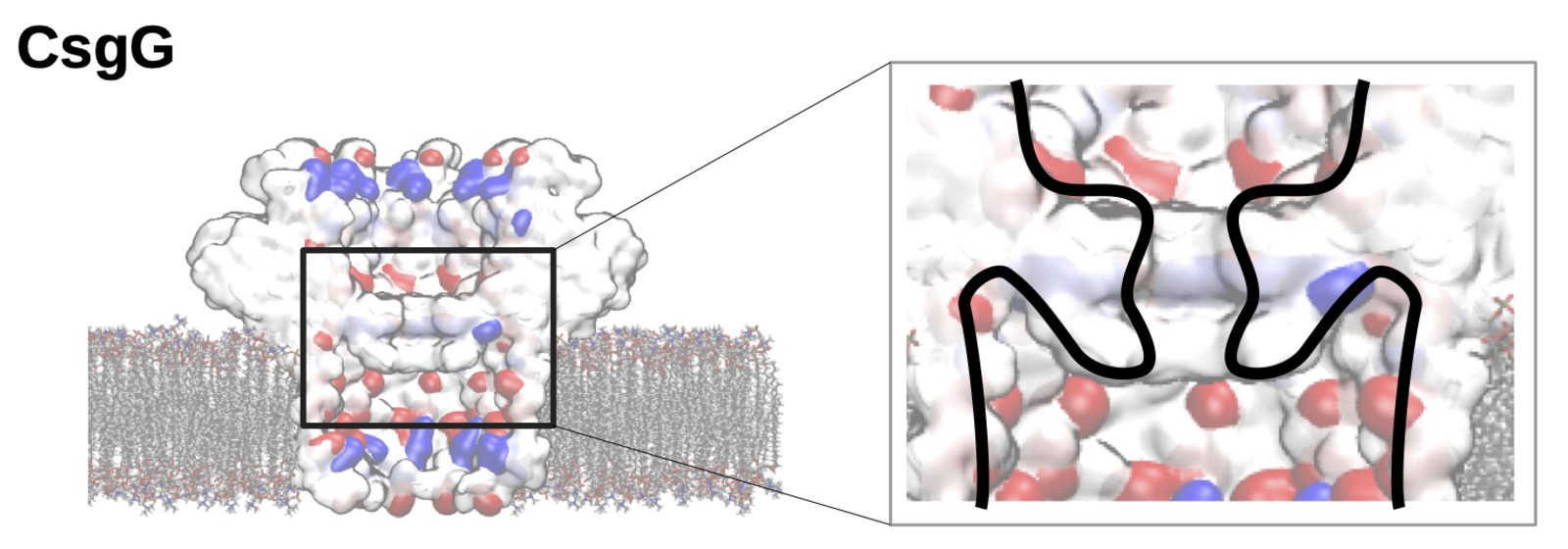
The team went on to examine this process using a biological nanopore called CsgG, which is already in use in commercial DNA sequencing kits. In addition, CsgG already possesses an ion-selective cavity next to its narrow sensing section. The simulations revealed that, indeed, the electroosmotic flow in this biological example is induced via this ion trapping cavity that is separated and independent from the sensing pore itself. “If these findings can be confirmed in experiments, we have found a way to decouple the capture of the protein to be sequenced from the capability to distinguish between amino acids — a huge advantage for the engineering of the nanopore,” explains Chinappi.
These results on nanopore protein sequencing can easily be generalized to other pores. Taken together, the team’s simulations suggest that biological nanopores like CsgG and α-hemolysin and are extremely promising and should be further pursued as a device for protein sequencing.
Image above: The nanopore CsgG is a possible sensor for future sequencing of single-protein molecules. The cavity surrounding the sensing section of the pore can, according to Mauro Chinappi’s simulations, induce a selective water flow that may bring protein molecules into the pore. (Image: M. Chinappi)
References:
- Di Mucco G., Morozzo della Rocca B. and Chinappi M.: Geometrical Induced Selectivity and Unifirectional Electroosmosis in Uncharged Nanopores. (in review).
- Alfaro J.A., Bohländer P., Dai M. et al.: The emerging landscape of single-molecule protein sequencing technologies. Nat Methods (2021). DOI: https://doi.org/10.1038/s41592-021-01143-1
- Di Muccio G., Rossini A.E., Di Marino D., Zollo G. and Chinappi M.: Insights into protein sequencing with an α-hemolysin nanopore by atomistic simulations. Sci Rep (2019). DOI: https://doi.org/10.1038/s41598-019-42867-7
- Bonome E.L., Cecconi F. and Chinappi M.: Electroosmotic flow through an a-hemolysin nanopore. Microfluid Nanofluidics (2017). DOI: https://doi.org/10.1007/s10404-017-1928-1
This article may be used on other media and online portals provided the copyright conditions are observed.