June 19, 2026 – by Santina Russo
It was the decisive breakthrough: in 2020, DeepMind launched AlphaFold 2, a machine learning model for predicting protein structures. For the first time, protein structures predicted directly from their amino acid sequence came close to the accuracy of structures determined experimentally. Only a little later, protein language models (pLMs) emerged as a parallel branch of protein prediction. These models learn directly from amino acid sequences and can predict sequence-structure-function relationships or generate new sequences. Together, these developments accelerate medical research, including the discovery of new drugs.
However, both approaches—AlphaFold’s structure prediction and pLM’s learning from sequences—struggle with predicting antibodies and their binding specificity. Antibodies are central to drug discovery. They are a major class of therapeutics themselves, and also critical tools that support the medical pipeline from protein target to diagnostics. Now, Sai Reddy, professor at ETH Zürich and Scientific Director of the Botnar Institute of Immune Engineering (BIIE), has developed a new neural network architecture to predict antibody-antigen binding—and, in the process, may have found a promising new way to predict many other medically relevant molecular binding pairs.
A collaboration for the common good
The work is the first outcome of a cooperation between BIIE and CSCS that started in summer 2025. “With this collaboration, we at CSCS wanted to support applied medical research for the public good,” says Joost VandeVondele, Deputy Director for Science at CSCS. BIIE’s aim is to tackle some of the most challenging and pressing problems in immune engineering—especially those that are not addressed by pharmaceutical companies. CSCS wants to help with this, says VandeVondele. “In the last few years, access to AI has broadened dramatically, and it has become much easier for domain specialists, such as the immunologists at BIIE, to build AI models. We wanted to contribute to this by making our supercomputing resources and experience available.”
Together with his team at BIIE, colleagues from ETH Zürich’s Department of Biosystems Science and Engineering, and supported by CSCS engineers, Reddy developed what he calls a Specificity Foundation Model—a language model designed to predicting antibody-antigen binding and its specificity directly from amino acid sequences.
Creating a thermodynamic binding map
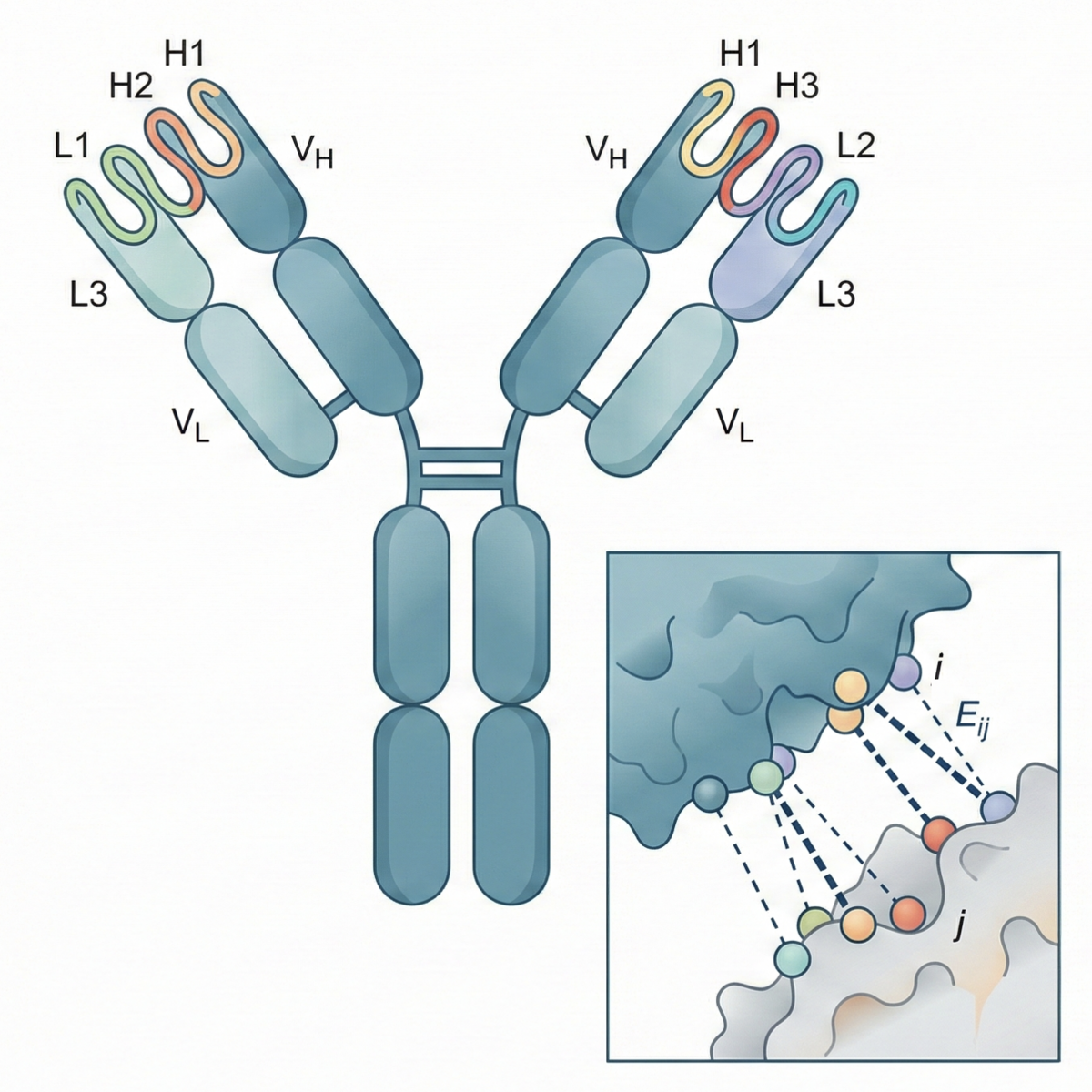
Most previous prediction models take an epitope as their starting point: a specific region of an antigen—usually a protein—that an antibody recognizes and binds to. From there, they propose likely binders. Reddy’s Specificity Foundation Model (SFM) takes a broader approach. It treats antibody-antigen binding as a thermodynamic recognition problem rather than as a structure-prediction problem. This is also why it works in both directions: given an antibody sequence, it retrieves a likely antigen or epitope, and vice versa.
The Botnar Institute: applied medical advances for the common good
The Botnar Institute of Immune Engineering (BIIE) is a new independent research institute founded in 2024 in Basel, with financial support from Fondation Botnar. Its purpose is to develop computational tools and immune-based solutions for the diagnosis, treatment, and prevention of diseases. Its research is meant to make advances in immune engineering and further develop them into practical medical applications, with a special focus on improving the health of children and adolescents worldwide.
To do this, the model maps antibodies and antigens into a joint embedding space. Simply put, the SFM converts antibody and antigen sequences into numerical vectors and places them in the same coordinate system. The model is then trained using contrastive learning, meaning that it learns from positive and negative pairs. In this case, a positive pair is an antibody and an antigen known to bind to each other; a negative pair is one in which they are known not to interact. In this way, the model creates a kind of binding map. Given a specific antigen, it can look for the nearest antibody, and vice versa.
An unusual data efficiency
With this approach, the antibody-antigen SFM achieved retrieval rates well above random: In about five out of ten cases, the correct binding partner was among the top ten. “While this means that it is not yet a prediction tool that can stand on its own, it gives strong suggestions,” says Reddy.
The work’s most significant result, however, may be how efficiently the model worked with the amount of data it was trained on—an exceedingly small dataset, in fact. It consisted of the amino acid sequences of roughly 3,400 antibody–antigen pairs. “Normally, for training a deep learning model, you would need millions to hundreds of millions of data points,” Reddy says. According to him, the data efficiency achieved with the SFM is roughly 100,000-fold greater than that of comparable contrastive learning architectures.
After investigating, Reddy found an explanation for the model’s unusual efficiency. It has to do with the similarity between the molecular binding energy landscape and the softmax attention mechanism built into today’s machine learning architectures. Molecule binding is described by the Boltzmann distribution, which, roughly put, states that the more energetically stable a bound state is relative to the free state of the binding partners, the higher the probability that the molecules will be bound at thermal equilibrium. In a similar way, the softmax mechanism turns raw similarity scores between tokens into probabilities. “In fact, these two mathematical equations that were developed independently from each other in different fields are identical,” says Reddy. “It’s this mathematical identity, which combines the physical conditions of molecular binding with the model’s architecture, that makes its learning so efficient.”
Applicable to any molecular binding?
This is also why Reddy says the same principle can be applied to many kinds of molecular binding—whether antibody to antigen, transcription factor to DNA, enzyme to substrate, drug candidate to protein target, and more. Reddy has specified SFM versions for ten such molecular binding regimes. “They all use the same AI architecture, as all these binding regimes are subject to the same thermodynamics represented by the Boltzmann distribution,” Reddy explains.
Most recently, he built, trained, and tested the drug-candidate-to-protein-target version, called dtSFM (drug-target Specificity Foundation Model) using CSCS’s Alps supercomputer. The model was trained on roughly 700,000 experimentally measured drug-protein interactions. According to the results reported in Reddy’s preprint paper, dtSFM showed strong performance. In nine out of ten cases, the model ranked the correct partner among the first ten hits.
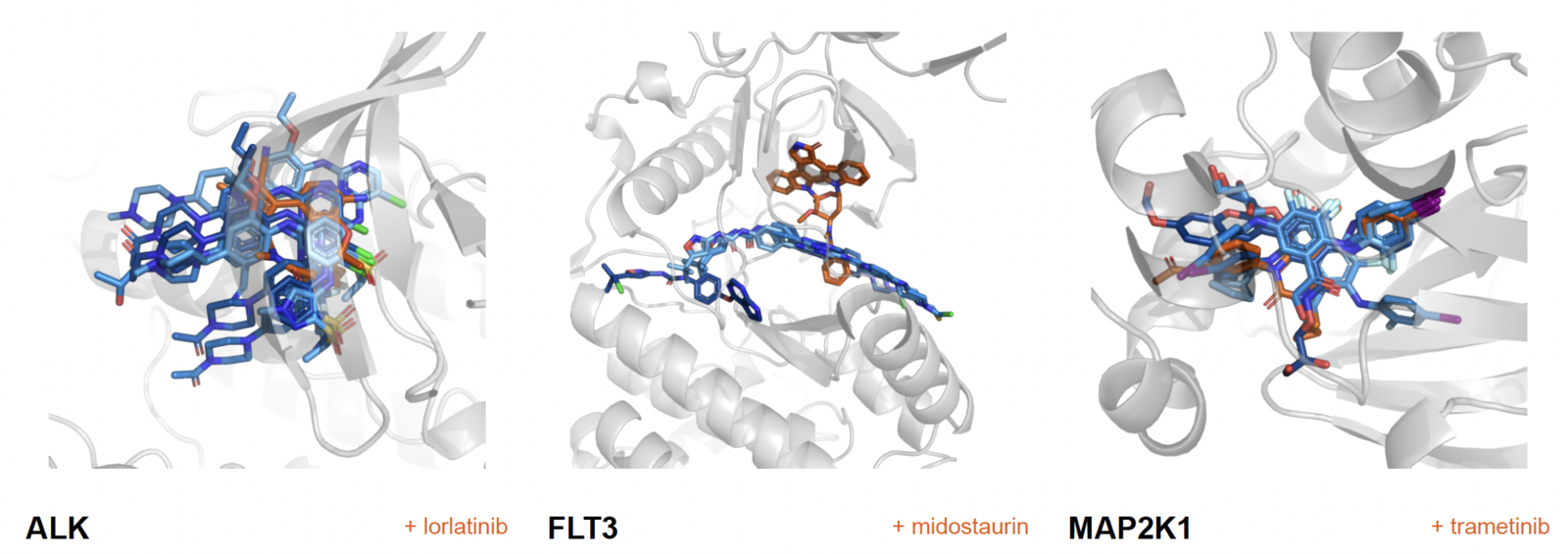
In addition, dtSFM is also the first of Reddy’s Specificity Foundation Models to include a generative function, meaning that it can generate new candidate molecules for a given target. In the reported results, the model generated molecules for 16 targets, all of them medically relevant. These target-drug pairs were then independently checked by structure prediction using AlphaFold 3. The result: of the 1,200 drug candidates designed by dtSFM, 850, or 71 percent, matched the AlphaFold 3 structural confidence of an approved drug.
It is important to note that while these results are promising, they are not yet peer-reviewed and still need to be validated experimentally in the lab. If the results turn out to be transferable to the wet lab and to clinical studies, however, they would suggest that drug candidates could be predicted and designed more directly and cost-efficiently than to date. In the long run, this could even help democratize the drug discovery process and enable organizations in under-resourced countries to improve the health and well-being of local populations.
References:
H. Lee, K. Castro, S. Renwick, L. Stalder, W. Glänzer, R. Kumar, N. Chen, A. Scheck, A. Yermanos, D. Mason, S.T. Reddy: Contrastive learning for antibody-antigen sequence-to-specificity prediction. Preprint (2026). DOI: https://doi.org/10.64898/2026.02.25.707916
S.T. Reddy: Computational Convergence of Adaptive Immunity and Artificial Intelligence. Preprint (2026). DOI: https://doi.org/10.64898/2026.02.03.703525
S.T. Reddy: Methods for Molecular Recognition Computing. Preprint (2026). DOI: https://doi.org/10.64898/2026.04.03.716328
S.T. Reddy: A Drug–Target Specificity Foundation Model for Off-target Prediction, Repurposing, and Generative Design. Preprint (2026). DOI: https://doi.org/10.64898/2026.06.08.730844
S.T. Reddy: Generative Drug Design in a Loop with dtSFM. Preprint (2026). DOI: https://doi.org/10.64898/2026.06.10.731501
Cover image: An illustration of antibody-antigen binding (Adobe Stock)